
Selection and Drift: A Comparison between Historic and Recent Dutch Friesian Cattle and Recent Holstein Friesian Using WGS Data


Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Animals
2.2. Short Read Sequencing Mapping and Variant Calling
2.3. Group Structure and Identification of Group-Specific SNPs
2.4. Genetic Diversity Parameters
2.5. Selection Signature Analysis
2.6. Measure of Runs of Homozygosity
2.7. Runs of Homozygosity Islands
3. Results
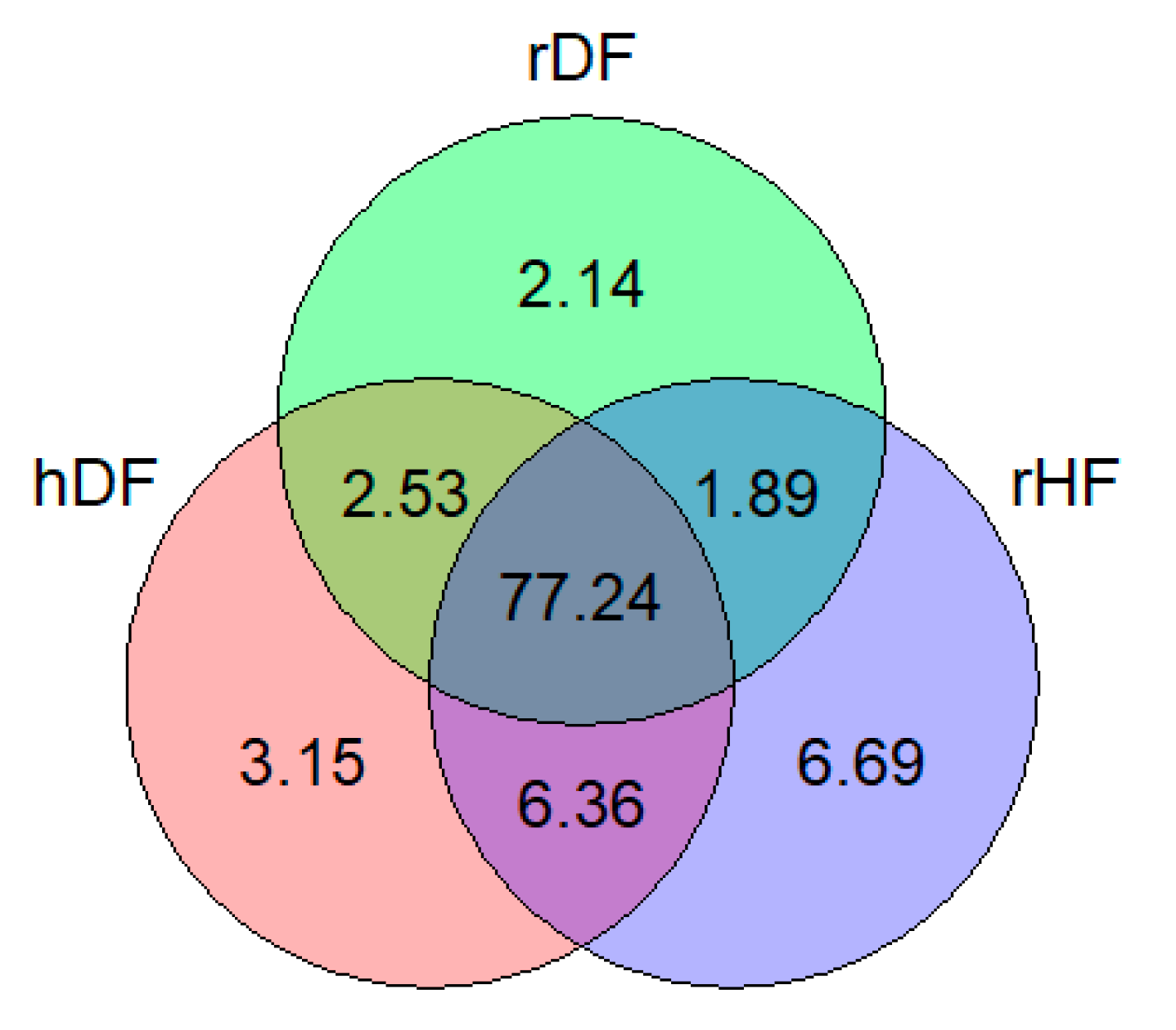
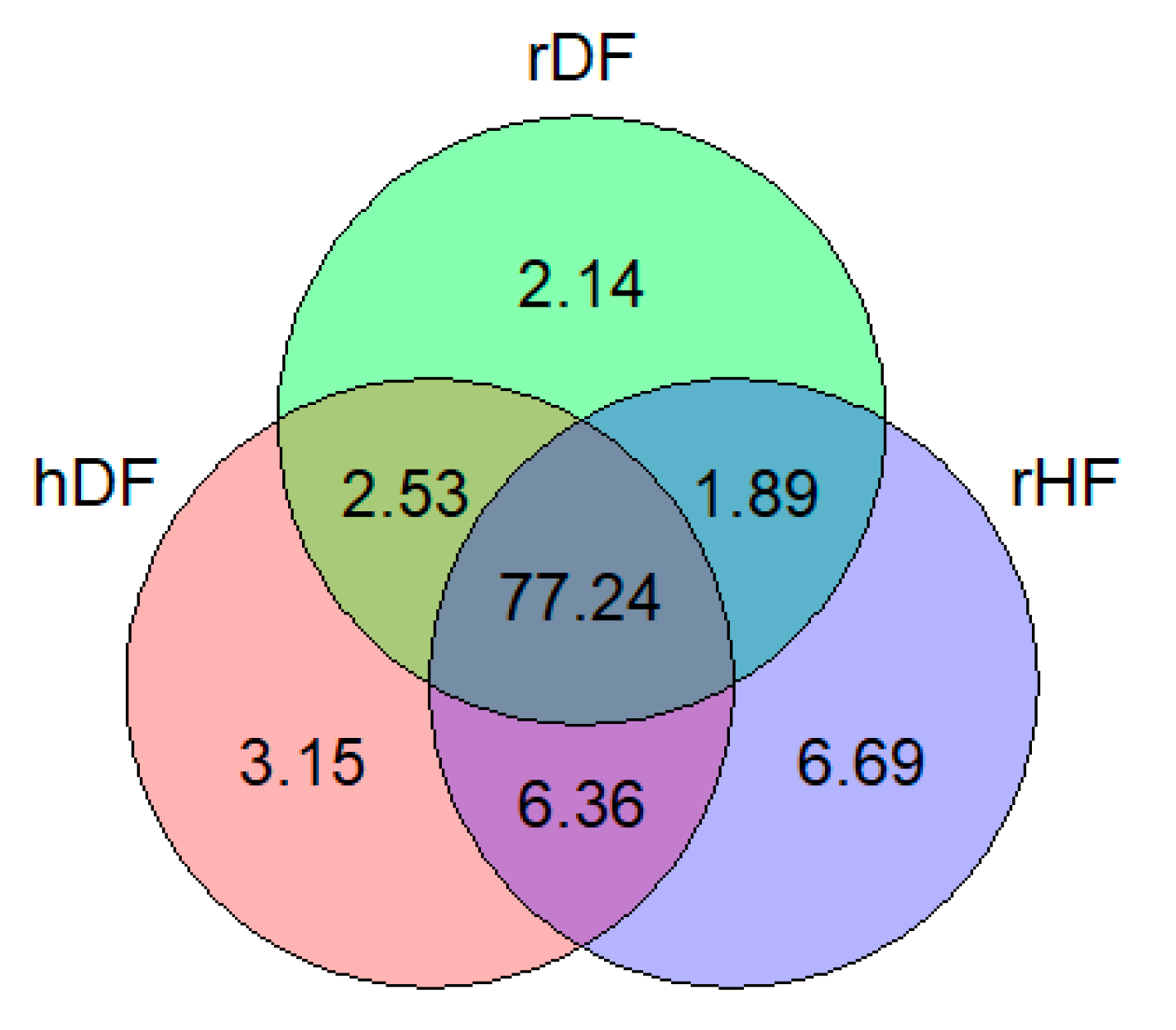
3.1. SNP Distribution
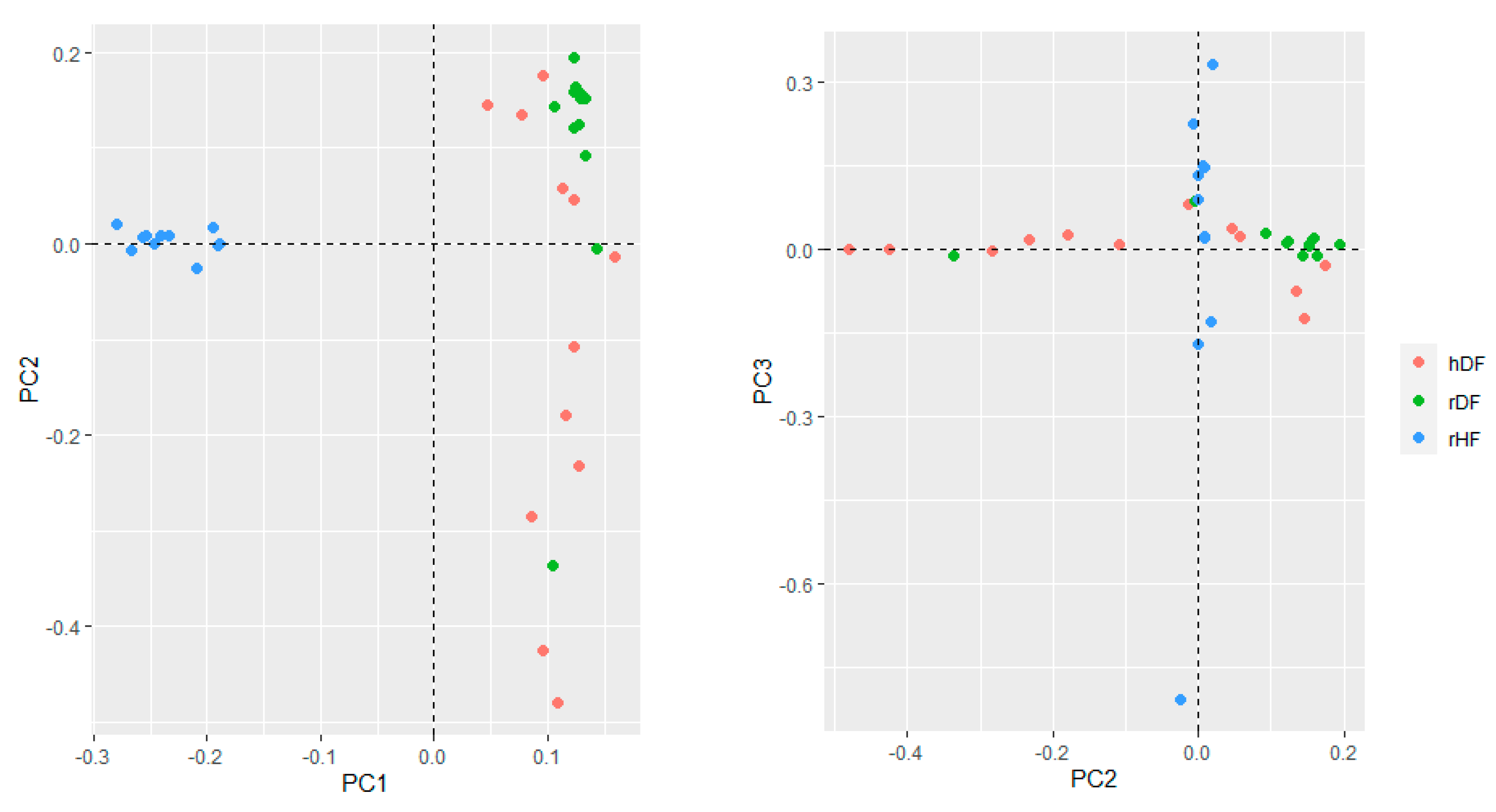
3.2. Genetic Diversity Parameters
3.3. Genomic Inbreeding Coefficients
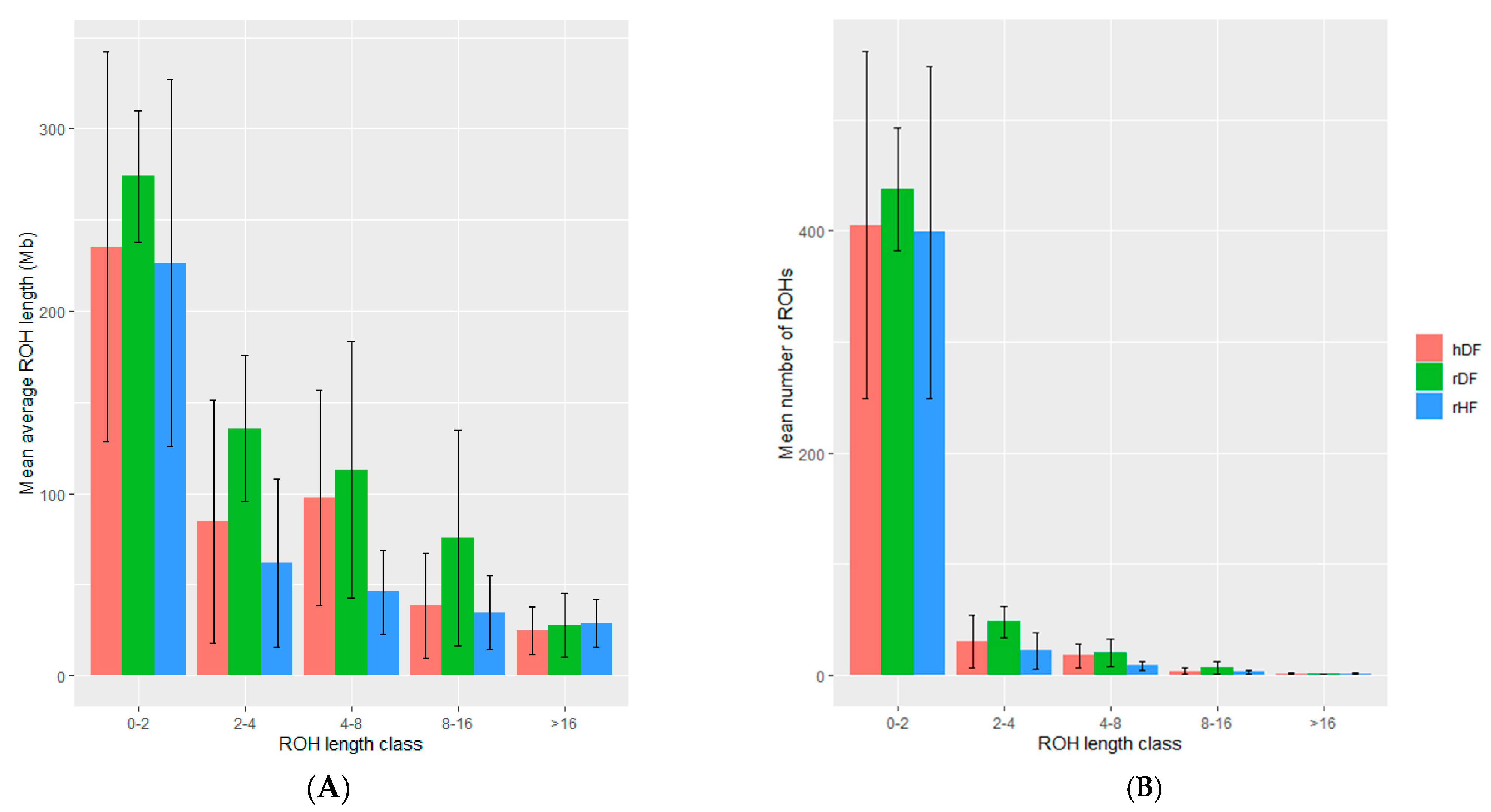
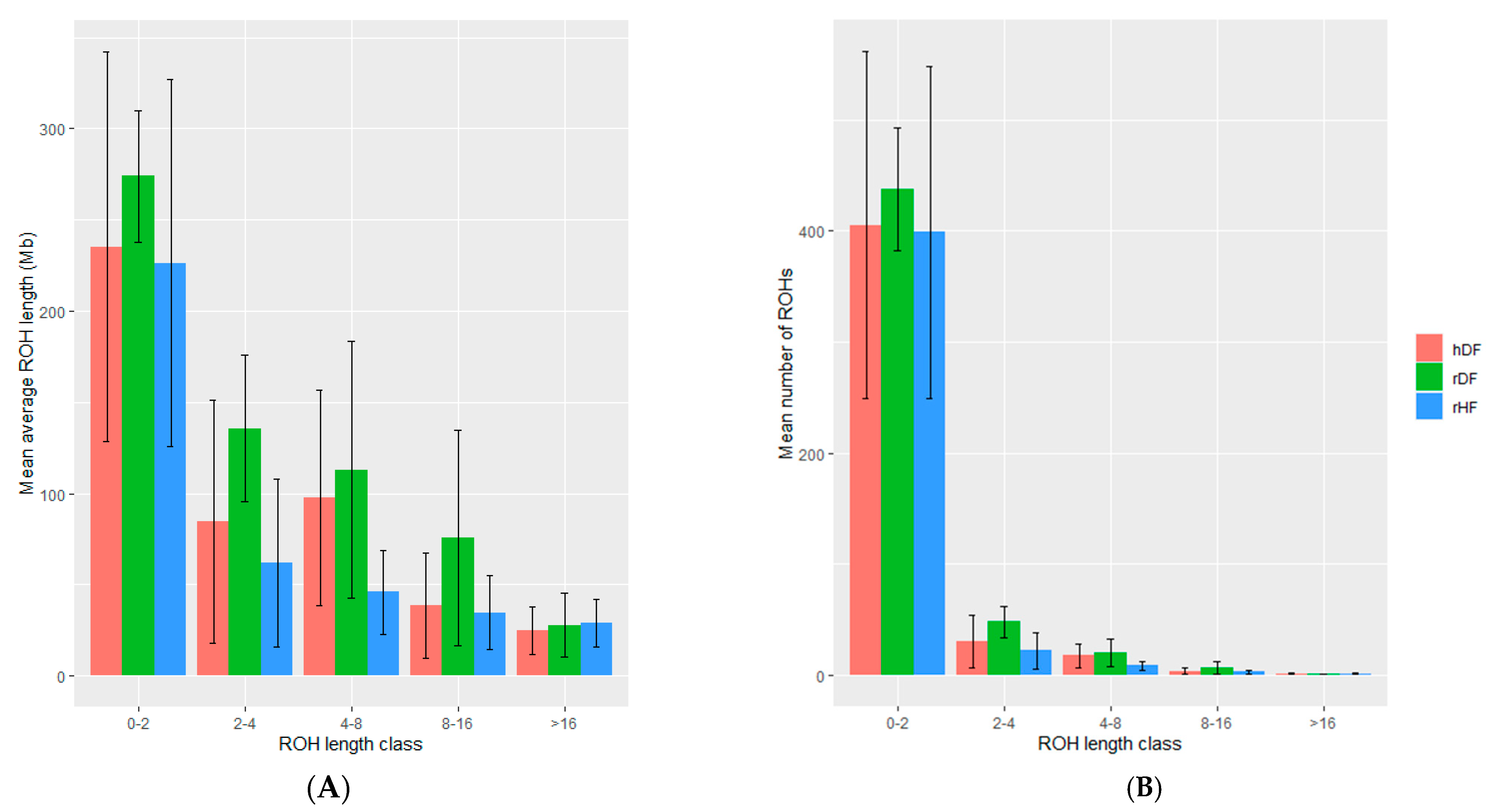
3.4. Measure of Runs of Homozygosity
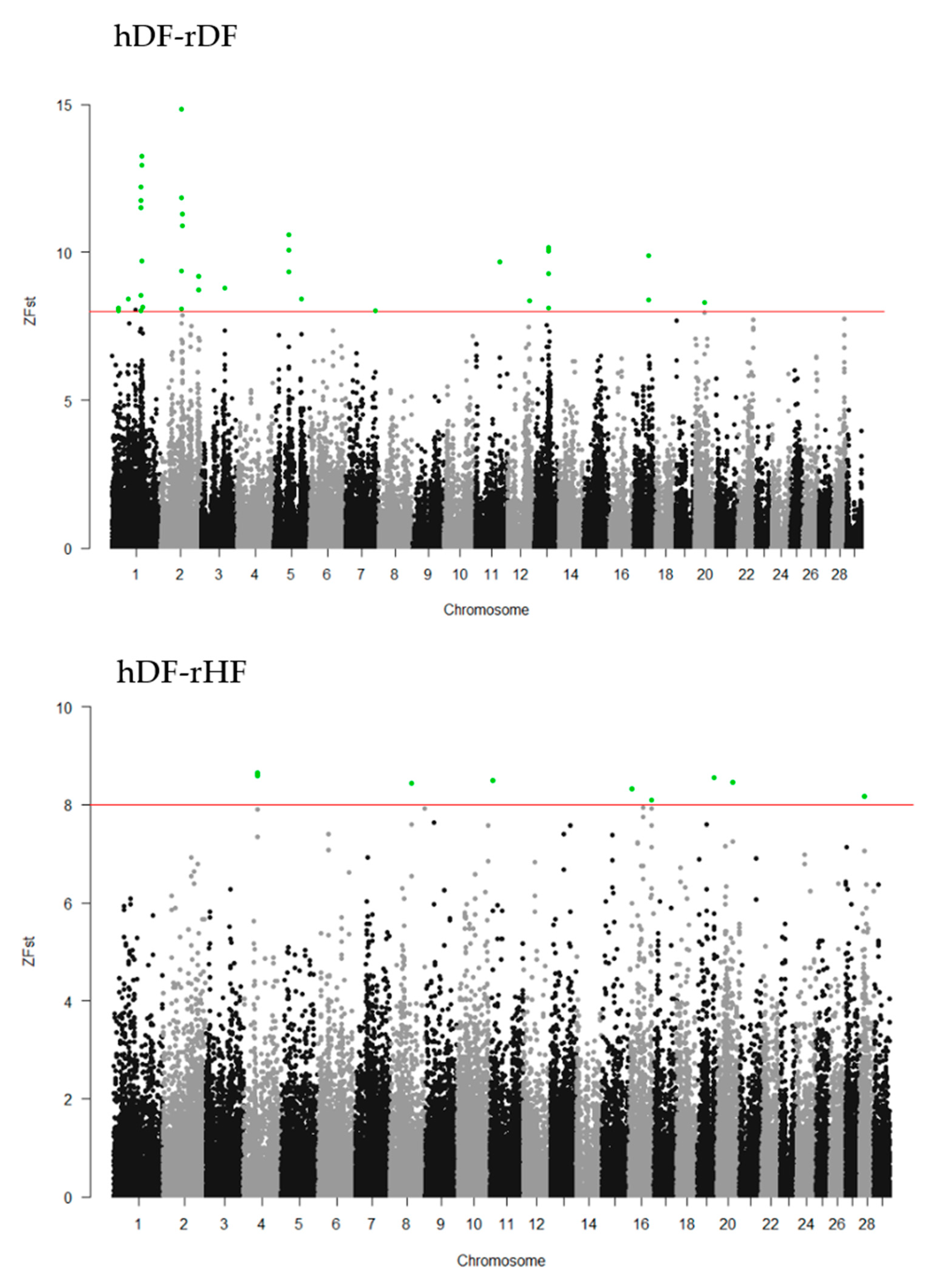
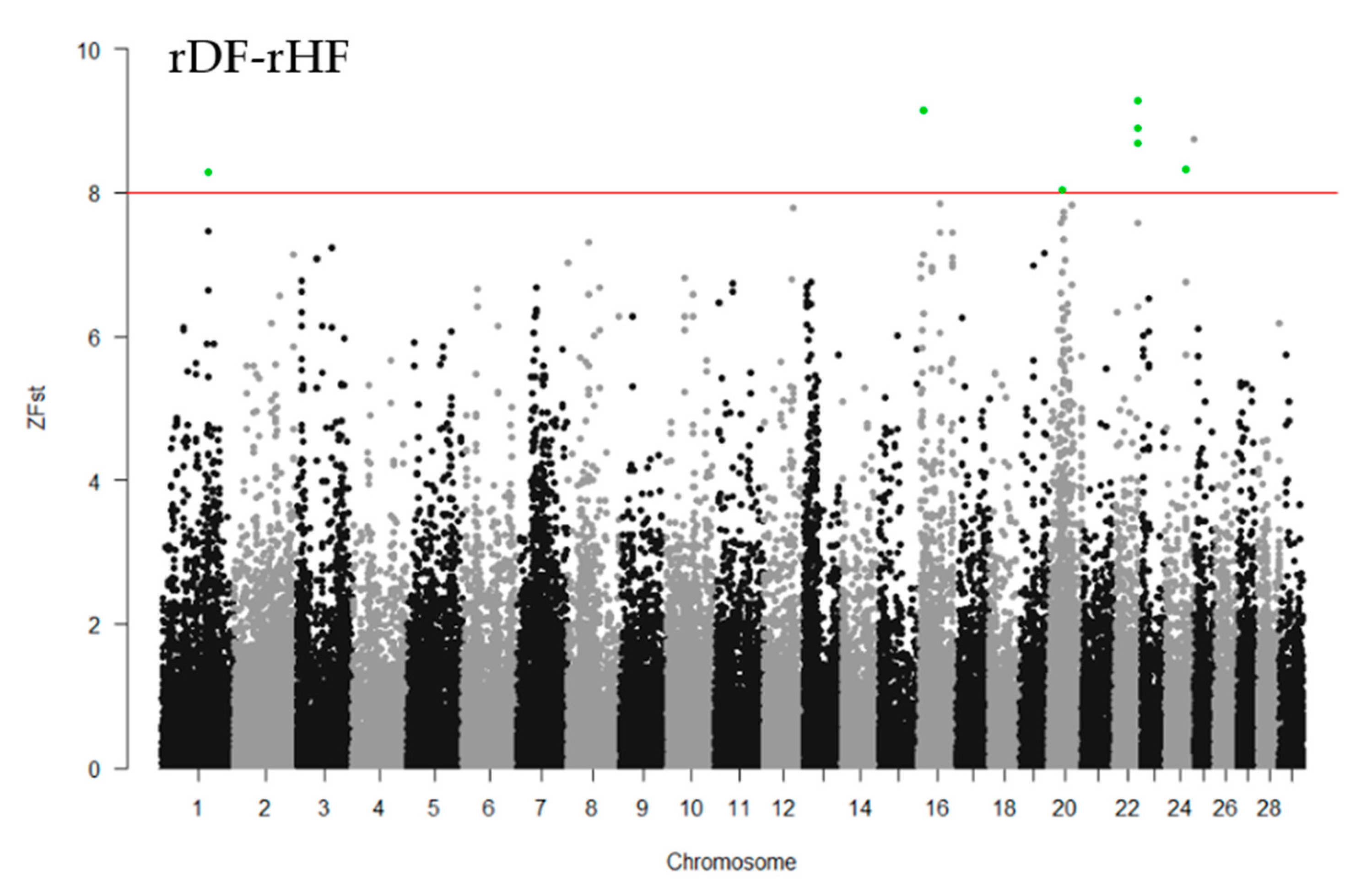
3.5. Genomewide Selection Signature Analysis
3.6. Runs of Homozygosity Islands
4. Discussion
4.1. General
4.2. Divergence between Groups
4.3. Genetic Diversity within Groups
4.4. Differentiated Genomic Regions
4.5. Runs of Homozygosity Detection and Distributions
4.6. Gene Bank
4.7. Limitation of the Study
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Marchitelli, C.; Consortium, E. Marker-assisted conservation of European cattle breeds: An evaluation. Anim. Genet. 2006, 37, 475. [Google Scholar]
- Medugorac, I.; Medugorac, A.; Russ, I.; Veit-Kensch, C.E.; Taberlet, P.; Luntz, B.; Mix, H.M.; Förster, M. Genetic diversity of European cattle breeds highlights the conservation value of traditional unselected breeds with high effective population size. Mol. Ecol. 2009, 18, 3394–3410. [Google Scholar] [CrossRef] [PubMed]
- Moscarelli, A.; Sardina, M.T.; Cassandro, M.; Ciani, E.; Pilla, F.; Senczuk, G.; Portolano, B.; Mastrangelo, S. Genome-wide assessment of diversity and differentiation between original and modern Brown cattle populations. Anim. Genet. 2021, 52, 21–31. [Google Scholar] [CrossRef] [PubMed]
- Hiemstra, S.J.; de Haas, Y.; Mäkit-Tanila, A.; Gandini, G. Local Cattle Breeds in Europe: Development of Policies and Strategies for Self-Sustaining Breeds; Wageningen Academic Publishers: Wageningen, The Netherlands, 2010. [Google Scholar]
- Oldenbroek, K. Utilisation and Conservation of Farm Animal Genetic Resources; Wageningen Academic Publishers: Wageningen, The Netherlands, 2007; pp. 1–232. [Google Scholar]
- Van Breukelen, A.E.; Doekes, H.P.; Windig, J.J.; Oldenbroek, K. Characterization of genetic diversity conserved in the gene bank for dutch cattle breeds. Diversity 2019, 11, 229. [Google Scholar] [CrossRef] [Green Version]
- Doekes, H.P. Genomic Characterization and Conservation of Genetic Diversity in Cattle; Wageningen University: Wageningen, The Netherlands, 2020. [Google Scholar]
- Theunissen, B. Breeding for nobility or for production?: Cultures of dairy cattle breeding in the Netherlands, 1945–1995. ISIS 2012, 103, 278–309. [Google Scholar] [CrossRef]
- Maurice-Van Eijndhoven, M.H.T. Genetic Variation of Milk Fatty Acid Composition between and within Dairy Cattle Breeds; Wageningen University: Wageningen, The Netherlands, 2014. [Google Scholar]
- Fimland, E.; Oldenbroek, K. Practical implications of utilisation and management. In Utilisation and Conservation of Farm Animal Genetic Resources; Wageningen Academic Publishers: Wageningen, The Netherlands, 2007; pp. 195–213. [Google Scholar]
- Pérez-Enciso, M.; Rincón, J.C.; Legarra, A. Sequence- vs. chip-assisted genomic selection: Accurate biological information is advised. Genet. Sel. Evol. 2015, 47, 43. [Google Scholar] [CrossRef] [Green Version]
- Eynard, S.E.; Windig, J.J.; Hiemstra, S.J.; Calus, M.P.L. Whole-genome sequence data uncover loss of genetic diversity due to selection. Genet. Sel. Evol. 2016, 48, 33. [Google Scholar] [CrossRef] [Green Version]
- Eusebi, P.G.; Martinez, A.; Cortes, O. Genomic tools for effective conservation of livestock breed diversity. Diversity 2020, 12, 8. [Google Scholar] [CrossRef] [Green Version]
- Hayes, B.J.; Daetwyler, H.D. 1000 Bull Genomes Project to Map Simple and Complex Genetic Traits in Cattle: Applications and Outcomes. Annu. Rev. Anim. Biosci. 2019, 7, 89–102. [Google Scholar] [CrossRef]
- Andrews, S. FASTQC. A quality control tool for high throughput sequence data. Fast QC 2010, 532, 1. [Google Scholar]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [Green Version]
- Okonechnikov, K.; Conesa, A.; García-Alcalde, F. Qualimap 2: Advanced multi-sample quality control for high-throughput sequencing data. Bioinformatics 2016, 32, 292–294. [Google Scholar] [CrossRef]
- Pirooznia, M.; Kramer, M.; Parla, J.; Goes, F.S.; Potash, J.B.; McCombie, W.R.; Zandi, P.P. Validation and assessment of variant calling pipelines for next-generation sequencing. Hum. Genom. 2014, 8, 14. [Google Scholar] [CrossRef] [Green Version]
- Jagt, C.V.; Chamberlain, A.; Schnabel, R.D.; Hayes, B.; Daetwyler, H. Which is the best variant caller for large whole-genome sequencing datasets? In Proceedings of the 11th World Congress on Genetics Applied to Livestock Production, Auckland, New Zealand, 11–16 February 2018.
- Ramos, A.M.; Megens, H.J.; Crooijmans, R.P.M.A.; Schook, L.B.; Groenen, M.A.M. Identification of high utility SNPs for population assignment and traceability purposes in the pig using high-throughput sequencing. Anim. Genet. 2011, 42, 613–620. [Google Scholar] [CrossRef]
- Purcell, S.; Neale, B.; Todd-Brown, K.; Thomas, L.; Ferreira, M.A.R.; Bender, D.; Maller, J.; Sklar, P.; De Bakker, P.I.W.; Daly, M.J.; et al. PLINK: A tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet. 2007, 81, 559–575. [Google Scholar] [CrossRef] [Green Version]
- Danecek, P.; Auton, A.; Abecasis, G.; Albers, C.A.; Banks, E.; DePristo, M.A.; Handsaker, R.E.; Lunter, G.; Marth, G.T.; Sherry, S.T.; et al. The variant call format and VCFtools. Bioinformatics 2011, 27, 2156–2158. [Google Scholar] [CrossRef]
- Weir, B.S.; Cockerham, C.C. Estimating F-statistics for the analysis of population structure. Evolution 1984, 38, 1358–1370. [Google Scholar] [CrossRef]
- Rafiepour, M.; Ebrahimie, E.; Vahidi, M.F.; Salekdeh, G.H.; Niazi, A.; Dadpasand, M.; Liang, D.; Si, J.; Ding, X.; Han, J.; et al. Whole-genome resequencing reveals adaptation prior to the divergence of buffalo subspecies. Genome Biolog. Evol. 2021, 13, evaa231. [Google Scholar] [CrossRef]
- Rubin, C.J.; Zody, M.C.; Eriksson, J.; Meadows, J.R.S.; Sherwood, E.; Webster, M.T.; Jiang, L.; Ingman, M.; Sharpe, T.; Ka, S.; et al. Whole-genome resequencing reveals loci under selection during chicken domestication. Nature 2010, 464, 587–591. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Turner, S.D. qqman: An R package for visualizing GWAS results using Q-Q and manhattan plots. bioRxiv 2014, 005165. [Google Scholar] [CrossRef]
- Cheng, J.; Zhao, H.; Chen, N.; Cao, X.; Hanif, Q.; Pi, L.; Hu, L.; Chaogetu, B.; Huang, Y.; Lan, X.; et al. Population structure, genetic diversity, and selective signature of Chaka sheep revealed by whole genome sequencing. BMC Genom. 2020, 21, 520. [Google Scholar] [CrossRef] [PubMed]
- Biscarini, F.; Cozzi, P.; Gaspa, G.; Marras, G. detectRUNS: Detect Runs of Homozygosity and Runs of Heterozygosity in Diploid Genomes. 2018. Available online: https://github.com/bioinformatics-ptp/detectRUNS/tree/master/detectRUNS (accessed on 3 May 2021).
- Gorssen, W.; Meyermans, R.; Buys, N.; Janssens, S. SNP genotypes reveal breed substructure, selection signatures and highly inbred regions in Piétrain pigs. Anim. Genet. 2020, 51, 32–42. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Felius, M.; Koolmees, P.A.; Theunissen, B.; Lenstra, J.A.; Baumung, R.; Manatrinon, S.; Mommens, G.; Holm, L.E.; Withen, K.B.; Pedersen, B.V.; et al. On the breeds of cattle-Historic and current classifications. Diversity 2011, 3, 660. [Google Scholar] [CrossRef] [Green Version]
- Hulsegge, I.; Schoon, M.; Windig, J.; Neuteboom, M.; Hiemstra, S.J.; Schurink, A. Development of a genetic tool for determining breed purity of cattle. Livest. Sci. 2019, 223, 60–67. [Google Scholar] [CrossRef]
- Upadhyay, M.; Eriksson, S.; Mikko, S.; Strandberg, E.; Stålhammar, H.; Groenen, M.A.M.; Crooijmans, R.P.M.A.; Andersson, G.; Johansson, A.M. Genomic relatedness and diversity of Swedish native cattle breeds. Genet. Sel. Evol. 2019, 51, 56. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hulsegge, B.; Calus, M.P.L.; Oldenbroek, J.K.; Windig, J.J. Conservation priorities for the different lines of Dutch Red and White Friesian cattle change when relationships with other breeds are taken into account. J. Anim. Breed. Genet. 2017, 134, 69–77. [Google Scholar] [CrossRef] [Green Version]
- CRV. Inbreeding in Dutch Cattle. 2021. Available online: https://www.cooperatie-crv.nl/wp-content/uploads/2021/01/Inbreeding--Dutch-Cattle-1.Pdf (accessed on 5 November 2021).
- Mill, R.; Nauta, W. Fundamentfokkerij Fries Hollands Vee; Louis Bolk Instituut: Driebergen, The Netherlands, 2010. [Google Scholar]
- Theunissen, B. A beautiful cow is a good cow. Dutch scientists and commercial breeders on cattle breeding, 1900–1950. Studium 2008, 1, 47–61. [Google Scholar] [CrossRef] [Green Version]
- Vohra, V.; Chhotaray, S.; Gowane, G.; Alex, R.; Mukherjee, A.; Verma, A.; Deb, S.M. Genome-Wide Association Studies in Indian Buffalo Revealed Genomic Regions for Lactation and Fertility. Front. Genet. 2021, 12, 696109. [Google Scholar] [CrossRef]
- Christensen, G.L.; Ivanov, I.P.; Atkins, J.F.; Mielnik, A.; Schlegel, P.N.; Carrell, D.T. Screening the SPO11 and EIF5A2 genes in a population of infertile men. Fertil. Steril. 2005, 84, 758–760. [Google Scholar] [CrossRef] [PubMed]
- Paul, N.; Kumaresan, A.; Das Gupta, M.; Nag, P.; Guvvala, P.R.; Kuntareddi, C.; Sharma, A.; Selvaraju, S.; Datta, T.K. Transcriptomic Profiling of Buffalo Spermatozoa Reveals Dysregulation of Functionally Relevant mRNAs in Low-Fertile Bulls. Front. Vet. Sci. 2021, 7, 609518. [Google Scholar] [CrossRef] [PubMed]
- Zaborski, D.; Grzesiak, W.; Pilarczyk, R. Detection of difficult calvings in the Polish Holstein-Friesian Black-and-White heifers. J. Appl. Anim. Res. 2016, 44, 42–53. [Google Scholar] [CrossRef]
- Cole, J.B.; Waurich, B.; Wensch-Dorendorf, M.; Bickhart, D.M.; Swalve, H.H. A genome-wide association study of calf birth weight in Holstein cattle using single nucleotide polymorphisms and phenotypes predicted from auxiliary traits. J. Dairy Sci. 2014, 97, 3156–3172. [Google Scholar] [CrossRef] [Green Version]
- CRV. Genetische Trends van Koeien in Nederland. 2021. Available online: https://cooperatiecrv-be6.kxcdn.com/wp-content/uploads/2021/08/gen_trend_koe_nl_20210819.pdf (accessed on 22 October 2021).
- Cerri, R.L.A.; Thompson, I.M.; Kim, I.H.; Ealy, A.D.; Hansen, P.J.; Staples, C.R.; Li, J.L.; Santos, J.E.P.; Thatcher, W.W. Effects of lactation and pregnancy on gene expression of endometrium of Holstein cows at day 17 of the estrous cycle or pregnancy. J. Dairy Sci. 2012, 95, 5657–5675. [Google Scholar] [CrossRef]
- Lonergan, P.; Fair, T.; Forde, N.; Rizos, D. Embryo development in dairy cattle. Theriogenology 2016, 86, 270–277. [Google Scholar] [CrossRef]
- Terenina, E.; Bazovkina, D.; Rousseau, S.; Salin, F.; D’Eath, R.; Turner, S.; Kulikov, A.; Mormede, P. Gene polymorphisms associated with aggression in pigs. Journées Rech. Porc. Fr. 2012, 44, 45–46. [Google Scholar]
- Zinovieva, N.A.; Dotsev, A.V.; Sermyagin, A.A.; Deniskova, T.E.; Abdelmanova, A.S.; Kharzinova, V.R.; Sölkner, J.; Reyer, H.; Wimmers, K.; Brem, G. Selection signatures in two oldest Russian native cattle breeds revealed using high-density single nucleotide polymorphism analysis. PLoS ONE 2020, 15, e0242200. [Google Scholar] [CrossRef]
- Smith, J.L.; Wilson, M.L.; Nilson, S.M.; Rowan, T.N.; Oldeschulte, D.L.; Schnabel, R.D.; Decker, J.E.; Seabury, C.M. Genome-wide association and genotype by environment interactions for growth traits in U.S. Gelbvieh cattle. BMC Genom. 2019, 20, 926. [Google Scholar] [CrossRef] [Green Version]
- An, B.; Xia, J.; Chang, T.; Wang, X.; Xu, L.; Zhang, L.; Gao, X.; Chen, Y.; Li, J.; Gao, H. Genome-wide association study reveals candidate genes associated with body measurement traits in Chinese Wagyu beef cattle. Anim. Genet. 2019, 50, 386–390. [Google Scholar] [CrossRef]
- Nishimura, S.; Watanabe, T.; Mizoshita, K.; Tatsuda, K.; Fujita, T.; Watanabe, N.; Sugimoto, Y.; Takasuga, A. Genome-wide association study identified three major QTL for carcass weight including the PLAG1-CHCHD7 QTN for stature in Japanese Black cattle. BMC Genet. 2012, 13, 40. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bouwman, A.C.; Daetwyler, H.D.; Chamberlain, A.J.; Ponce, C.H.; Sargolzaei, M.; Schenkel, F.S.; Sahana, G.; Govignon-Gion, A.; Boitard, S.; Dolezal, M.; et al. Meta-analysis of genome-wide association studies for cattle stature identifies common genes that regulate body size in mammals. Nat. Genet. 2018, 50, 362–367. [Google Scholar] [CrossRef] [PubMed]
- Sölkner, J.; Ferencakovic, M.; Karimi, Z.; OBrien, A.M.P.; Mészáros, G.; Eaglen, S.A.E.; Boison, S.A.; Curik, I. Extremely Non-uniform: Patterns of Runs of Homozygosity in Bovine Populations. In Proceedings of the 10th World Congress of Genetics Applied to Livestock Production, Vancouver, BC, Canada, 17–22 August 2014. [Google Scholar] [CrossRef]
- Xu, L.; Zhao, G.; Yang, L.; Zhu, B.; Chen, Y.; Zhang, L.; Gao, X.; Gao, H.; Liu, G.E.; Li, J. Genomic Patterns of Homozygosity in Chinese Local Cattle. Sci. Rep. 2019, 9, 16977. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jang, J.; Kim, K.; Lee, Y.H.; Kim, H. Population differentiated copy number variation of Bos taurus, Bos indicus and their African hybrids. BMC Genom. 2021, 22, 531. [Google Scholar] [CrossRef] [PubMed]
- Bongiorni, S.; Gruber, C.E.M.; Bueno, S.; Chillemi, G.; Ferrè, F.; Failla, S.; Moioli, B.; Valentini, A. Transcriptomic investigation of meat tenderness in two Italian cattle breeds. Anim. Genet. 2016, 47, 273–287. [Google Scholar] [CrossRef]
- Júnior, G.A.F.; Costa, R.B.; De Camargo, G.M.F.; Carvalheiro, R.; Rosa, G.J.M.; Baldi, F.; Garcia, D.A.; Gordo, D.G.M.; Espigolan, R.; Takada, L.; et al. Genome scan for postmortem carcass traits in nellore cattle. J. Anim. Sci. 2016, 94, 4087–4095. [Google Scholar] [CrossRef]
- Hlongwane, N.L.; Hadebe, K.; Soma, P.; Dzomba, E.F.; Muchadeyi, F.C. Genome Wide Assessment of Genetic Variation and Population Distinctiveness of the Pig Family in South Africa. Front. Genet. 2020, 11, 344. [Google Scholar] [CrossRef]
- Paiva, S.R.; McManus, C.M.; Blackburn, H. Conservation of animal genetic resources—A new tact. Livest. Sci. 2016, 193, 32–38. [Google Scholar] [CrossRef]
- Blackburn, H.D. Genetic selection and conservation of genetic diversity. Reprod. Domest. Anim. 2012, 47, 249–254. [Google Scholar] [CrossRef]
- Boitard, S.; Paris, C.; Sevane, N.; Servin, B.; Bazi-Kabbaj, K.; Dunner, S. Gene Banks as Reservoirs to Detect Recent Selection: The Example of the Asturiana de los Valles Bovine Breed. Front. Genet. 2021, 12, 575405. [Google Scholar] [CrossRef]
- Hulsegge, I.; Calus, M.; Hoving-Bolink, R.; Lopes, M.; Megens, H.J.; Oldenbroek, K. Impact of merging commercial breeding lines on the genetic diversity of Landrace pigs. Genet. Sel. Evol. 2019, 51, 60. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schmidt, T.L.; Jasper, M.; Weeks, A.R.; Hoffmann, A.A. Unbiased population heterozygosity estimates from genome-wide sequence data. Methods Ecol. Evol. 2021, 12, 1888–1898. [Google Scholar] [CrossRef]
- Nazareno, A.G.; Bemmels, J.B.; Dick, C.W.; Lohmann, L.G. Minimum sample sizes for population genomics: An empirical study from an Amazonian plant species. Mol. Ecol. Resour. 2017, 17, 1136–1147. [Google Scholar] [CrossRef]
- Willing, E.M.; Dreyer, C.; van Oosterhout, C. Estimates of genetic differentiation measured by fst do not necessarily require large sample sizes when using many snp markers. PLoS ONE 2012, 7, e42649. [Google Scholar] [CrossRef] [Green Version]
- Ahrens, C.W.; Rymer, P.D.; Stow, A.; Bragg, J.; Dillon, S.; Umbers, K.D.L.; Dudaniec, R.Y. The search for loci under selection: Trends, biases and progress. Mol. Ecol. 2018, 27, 1342–1356. [Google Scholar] [CrossRef] [Green Version]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Group | Abbreviation | MAF | Ho | He |
|---|---|---|---|---|
| Historic Dutch Friesian | hDF | 0.165 ± 0.152 a | 0.195 ± 0.025 | 0.250 ± 0.0005 a |
| Recent Dutch Friesian | rDF | 0.164 ± 0.155 b | 0.201 ± 0.015 | 0.250 ± 0.0003 a |
| Recent Holstein Friesian | rHF | 0.161 ± 0.153 c | 0.188 ± 0.035 | 0.249 ± 0.0016 b |
| hDF | rDF | rHF | |
|---|---|---|---|
| hDF | - | 0.0005 | 0.0624 |
| rDF | 0.0100 | - | 0.0719 |
| rHF | 0.0978 | 0.1105 | - |
| Froh | |||||
|---|---|---|---|---|---|
| Group | General Mean | ROH > 2 Mb | ROH > 4 Mb | ROH > 8 Mb | ROH > 16 Mb |
| Historic Dutch Friesian | 0.169 ± 0.095 ab (12) | 0.081 ± 0.061 ab (11) | 0.058 ± 0.034 (9) | 0.019 ± 0.012 (9) | 0.010 ± 0.005 (3) |
| Recent Dutch Friesian | 0.243 ± 0.062 a (12) | 0.132 ± 0.066 a (12) | 0.078 ± 0.055 (12) | 0.035 ± 0.028 (11) | 0.011 ± 0.007 (5) |
| Recent Dutch Friesian | 0.130 ± 0.067 b (12) | 0.047 ± 0.036 b (10) | 0.031 ± 0.022 (7) | 0.019 ± 0.013 (5) | 0.012 ± 0.005 (2) |
| Group | # Animals | Number of ROH | Total ROH Length (Mb) | Average ROH Length (Mb) | |
|---|---|---|---|---|---|
| Mean ± sd | Range | Mean ± sd | Mean ± sd | ||
| hDF | 12 | 449.75 ± 180.96 | 132–745 | 421.04 ± 235.43 ab | 0.87 ± 0.31 a |
| rDF | 12 | 513.50 ± 44.14 | 421–570 | 603.54 ± 154.92 a | 1.19 ± 0.34 b |
| rHF | 12 | 424.00 ± 161.48 | 75–653 | 323.92 ± 166.72 b | 0.73 ± 0.22 a |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hulsegge, I.; Oldenbroek, K.; Bouwman, A.; Veerkamp, R.; Windig, J. Selection and Drift: A Comparison between Historic and Recent Dutch Friesian Cattle and Recent Holstein Friesian Using WGS Data. Animals 2022, 12, 329. https://doi.org/10.3390/ani12030329
Hulsegge I, Oldenbroek K, Bouwman A, Veerkamp R, Windig J. Selection and Drift: A Comparison between Historic and Recent Dutch Friesian Cattle and Recent Holstein Friesian Using WGS Data. Animals. 2022; 12(3):329. https://doi.org/10.3390/ani12030329
Chicago/Turabian StyleHulsegge, Ina, Kor Oldenbroek, Aniek Bouwman, Roel Veerkamp, and Jack Windig. 2022. "Selection and Drift: A Comparison between Historic and Recent Dutch Friesian Cattle and Recent Holstein Friesian Using WGS Data" Animals 12, no. 3: 329. https://doi.org/10.3390/ani12030329
APA StyleHulsegge, I., Oldenbroek, K., Bouwman, A., Veerkamp, R., & Windig, J. (2022). Selection and Drift: A Comparison between Historic and Recent Dutch Friesian Cattle and Recent Holstein Friesian Using WGS Data. Animals, 12(3), 329. https://doi.org/10.3390/ani12030329